Applying for an NSF DDIG for the first time is an experience full of lots of emotions. I tried to put together some advice to prevent a few less tears being shed.
One does not just simply "submit" a DDIG. It is a several week-long process full of joy and laughter, and the only way to prevent a stress-induced stomach ulcer is to sprinkle the experience with sarcastic remarks. Here is a short guide for getting yourself set-up in FASTLANE and submitting to COEUS (Stony Brook's version of the grant-submitting interface that mirrors online forms of the 1990s).
A true and serious piece of advice is to begin this two weeks before the deadline (at least). Believe me, procrastination will do you no favors. Do this before you have a draft together. The minimum you need to get started is the title of your project (which you can always change later). But you need to get started. Also, make sure you have your IACUC and IRB approval taken care of well in advance, because you will need this for COEUS as well.
[1]
First, you must apply for an NSF FastLane login name. This is not something you sign up for on the NSF website, you have absolutely no control over this. Someone in your university will grant you this ID. This is different than the GFRP login if you are a GFRP fellow. Forget the beautifully designed FastLane submission form of the GFRP, where the sea-green trim borders all of the boxes and you actually may feel happy about life. Those days are in your past and you must now submit yourself to the harsh reality that the federal government does not have a budget for maintaining web submission forms post the early 2000s, and never will. To request your NSF ID (summarized in better detail
here), you must e-mail your Department Grant Officer. For Ecology & Evolution, it is (at the time of writing) Gloria (and she is truly great!).
[2]
Log in to FastLane. Go to
https://www.fastlane.nsf.gov, click "Proposals, Awards and Status". Using your new login information, login. Click "Proposal Functions", "Proposal Preparation", and "Prepare Proposal". Also, update any of your FastLane information. For a DDIG, your adviser (the PI) must do this first. Sit with them (or do it for them), with your NSF ID, and create the new proposal.
[3]
Log in to COEUS and begin a new proposal. Your adviser may have to do this first and then add you. You first must at least fill out the first page of creating a new proposal. Either do this for your adviser (before you do it for you) or sit with your adviser. Under the tab "Investigators/Key Persons", they can now add you as a Co-PI. As an added Co-PI, you need to fill out a series of questions to certify.
 |
| sunshine and happiness |
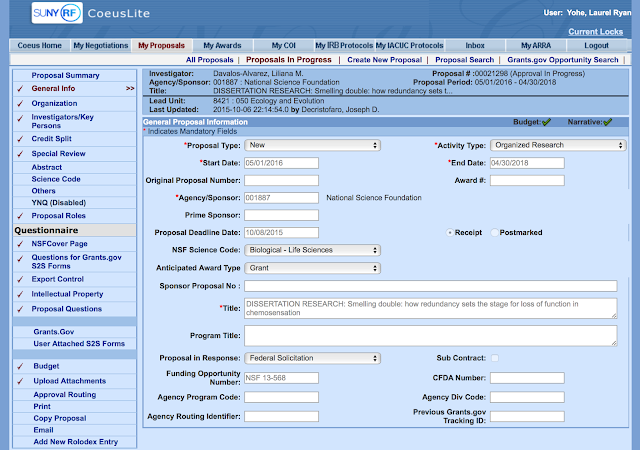 |
| An example of my login page |
Things to pay attention to-- the start date for the NSF DEB begins at the earliest 05/01 of the next year following submission and can last for 24 months. Note that the end date could not be 05/01/2018, but must be 04/30/2018. The title must begin with "DISSERTATION RESERCH:......".
[4] Add the Grants Management Officer (in this case, Gloria) as an aggregator. Before you yourself can do anything, the PI (your adviser) must give you permission to add and edit the proposal. On the left, click on "Proposal Roles". Add yourself and Gloria as an aggregator.
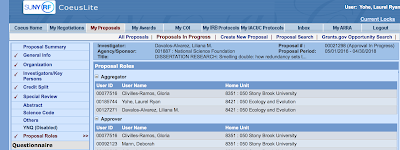 |
| You add the permissions for the people under Aggregator, the Grants Officer takes care of the rest |
[5] Fill out "Credit Split" tab. I really had no clue what was going on in this tab. Basically, give your adviser all of the credit and make sure the numbers add up to 100.
[6] Under the "Special Review" tab, upload any IACUC of IRB approvals associated with your project (there are certain numbers to report associated with your project.
[7] Update the "Investigators/Key Persons" tab to reflect the amount of "Effort" you will be putting into the project. Even though the DDIG has no salary associated with it, this still must be reported Remember that the university cares about nothing except money and it likes to know where it should be distributed over what frame of time, even if its not even there.
Notice that the C is referring to Calendar Year %Effort, not months. 1 month Calendar Year is 8.33% (1 / 12mo); 1 month AY 11.11% (1 / 9mo); 1 month summer 33.33% (1 / 3mo). My advisers effort is 1 month out of 12 calendar months. Mine is 11 out of 12 calendar months. This is where you can mess up because this effort MUST match what you also put in your CURRENT & PENDING documents. You can see my approval is still in progress, as an effect of how I did manage to mess this up. :-|
[8]Answer all the random questions in the tabs under "Questionnaire". I do recall these being relatively painless.
[9] Prepare the documents you need to submit to COEUS. Don't do anything in the budget tab. Instead, fill out the "Budget Worksheet", an excel file where you add everything and then upload it under "Upload Documents". There should be three tabs, each with different forms you need to update. These forms change all of the time so make sure from the COEUS forms tab, you are downloading the right ones. Some documents you can generate the template yourself.
An example of a successful DDIG with all of the parts can be found
here. Feel free to contact me if you would like more Stony Brook-related examples of some of these documents.
- Under the tab "Upload Proposal Documents"
- COEUS project summary: This can be the DDIG project summary format (Overview, Intellectual Merit, Broader Impacts) or if this is not ready yet, you can submit a random paragraph from your project that summarizes the broad objectives.
- DDIG Facilities statement: Basically, you summarize anything equipment, computing facilities, or laboratories you will be in your project. Make a statement about the Stony Brook Libraries too. See the example above for a model of (in my opinion, bare minimum) facilities page. I will be happy to provide an example of thorough one.
- DDIG Budget Justification: A one-two page summary that includes a timeline of how the money will be used in the proposal. Don't go super crazy (e.g. not necessary to include the exact quote for a Qiagen extraction kit), but at least have some estimates of each experiment in the objectives of your proposal, estimates of fieldwork costs, and . Also include an IDC statement in your description of Indirect Costs. (Literally copy and paste the following sentence: Foundation for SUNY at Stony Brook's most current approved rate agreement negotiated through DHHS, their federal cognizant agency, dated Feb. 26, 2015.) The Indirect Cost estimates can be calculated from the Budget Worksheet. See the PDF posted above for an example of this document.
- Under the "Upload Personnel Attachments" tab:
- Under the "Upload Institutional Attachments" tab:
- Internal COI document for EACH PI and Co-PI declaring any conflicts of interest, even if there are none. The more recent of this form is available on the COEUS website.
- COEUS proposal form: Similar to the information declared under "Investigators/Key Persons" tab. This information must also match, as well as what was under C&P. This form is available to download on the COEUS webpage.
- Budget Spreadsheet. This is a handy spreadsheet you can download from the COEUS webpage where you enter the values of your budget per year and then it estimates the Indirect Costs. This supplements doing anything under the Budget Tab in COEUS.
Once you have all of this uploaded to your COEUS, contact your Gloria-person and tell her you are ready to submit. She will check and tell you if you need to fix anything (you probably do, so this is why you need to start way in advance). Thinks like the Current & Pending, Biosketch, Budget Justification, and Facilities also need to be uploaded to FastLane. You need to fill out your budget on FastLane as well.
When you are getting ready to submit your DDIG (this day will come), you will have to "Allow SRO access" on your FastLane account so that the Gloria person can review and submit (no, you can't do that yourself). I hope you have come to the realization that you have so little control over your life.
Live, learn, endure. You can do it!





